Vibrational frequencies
A frequency calculation can be performed either numerically or analytically. The numerical approach is more genera and is implemented directly in ASH. This approach can always be applied to any QM or MM theory or hybrid theories like QM/MM, ONIOM and Wrap theories, as long as an accurate gradient (typically analytic) is available.
For some QM-programs it is also possible to request an analytical Hessian. This is only available for some QM programs and requires this features to be enabled in the ASH interface. Currently supported programs: ORCA, CFour, pyscf.
For hybrid theories like QM/MM Hamiltonians, the numerical approach is often the only available option.
Numerical frequencies
Numerical frequencies can be performed with ASH using any QM, MM or QM/MM theory object. A QM-method that has available an analytical gradient (i.e. 1st derivative of energy) in the external code should typically be used. Most HF, DFT, MP2 and CASSCF methods have analytical gradients implemented in QM codes.
Note that a numerical 2nd derivative on top of a numerical 1st derivative can in principle be done but is not recommended (accumulation of numerical noise).
Use the NumFreq function to request a numerical frequency job. The function requires a fragment object and a theory level at minimum. The fragment object should typically contain a fragment with optimized coordinates at same level of theory (i.e. an already optimized minimum or saddlepoint).
def NumFreq(fragment=None, theory=None, npoint=2, displacement=0.005, hessatoms=None, numcores=1, runmode='serial',
temp=298.15, pressure=1.0, hessatoms_masses=None, printlevel=1, charge=None, mult=None):
Keyword |
Type |
Default value |
Details |
|---|---|---|---|
|
ASH Fragment |
None |
ASH Fragment object. |
|
ASHTheory object |
None |
ASH Theory object. |
|
String |
'serial |
Whether to run the numerical frequency displacement calculations in serial (sequentially)
or in parallel using Python multiprocessing.
|
|
Integer |
2 |
Whether to do a 1-point or 2-point approximation for the Hessian. 1-point takes half as many
displacements as 2-point and is less accurate.
|
|
Float |
0.005 |
Size of displacement (in Å) to take. |
|
list |
None |
Optional list of atom indices that will be displaced and define a partial Hessian instead
of a full Hessian. If None, all atoms in Fragment will be displaced.
|
|
Float |
298.15 |
Temperature (in K) to use in thermochemistry calculation. |
|
Float |
1.0 |
Pressure (in atm) to use in thermochemistry calculation. |
|
list |
None |
Optional list of masses for the Hessian atoms. |
|
integer |
None |
Optional charge. Will override charge attribute of ASH Fragment. |
|
integer |
None |
Optional spin multiplicity. Will override mult attribute of ASH Fragment. |
|
integer |
1 |
The printlevel. |
1-point vs. 2-point Hessian
Additionally you can select to do a 1-point Hessian or a 2-point Hessian by the npoint keyword (value of 1 or 2). A 1-point Hessian makes a single displacement (+ direction) for each atom and each x,y and z-coordinate from the input geometry. This option is reasonably accurate and is the default. A more accurate 2-point Hessian makes displacement in both + and - directions (for each x-, y- and z-coordinate of each atom), is twice as expensive (double the displacements) but is more accurate. The displacement step can be changed if required. The default recommended setting is: 0.0005 Å.
Serial or parallel calculation
Two runmodes are available: 'serial' and 'parallel'.
Serial
The default 'serial' mode will run each displacement sequentially. The Energy+Gradient calculation of each displacment can, however, still be parallelized if the QM or QM/MM object has this information available. For example if an ORCATheory object has been defined with numcores=8 then ORCA will run each Energy+Gradient evaluation with 8 cores using the OpenMPI parallelization of ORCA.
Parallel
For numerical frequencies, it is usually much more efficient, however, to run the displacement jobs simultaneously in parallel fashion. This is accomplished using runmode='parallel' and the parallelization will be linear scaling (almost always recommended). As there are almost always many more displacements available than CPUs, the parallelization of the QM or QM/MM object should usually be turned off and instead as many displacements are run simultaneously as there are number of cores. For example, for a 30-atom system, there are 90 XYZ coordinates. For a 2-point Hessian, this means that 180 displacements to be calculated. If 20 cores are available, then 20 displacements can be run simultaneously, fully utilizing all 20 cores. This will require 9 runs in total (20*9=180).
Warning
Parallel runmode is currently not available for QM/MM calculations.
Full or partial Hessian
A partial Hessian can be easily performed instead of the full Hessian. This is an excellent approximation for vibrational modes with rather local character and the quality of the approximation can be controlled.
For a QM/MM model of a protein active site with a common active region of about 1000 atoms, the full Hessian of all 1000 atoms would actually not be a doable or recommended calculation. Instead a partial Hessian job of the important atoms (e.g. the QM region) makes more sense.
A partial Hessian job is performed if a list of Hessian atoms (e.g. hessatoms=[0,1,2] ) is passed to the NumFreq function. In this case, the displacements will only be calculated for the list of "hessatoms" and the result is a partial Hessian for the chosen atoms of the system.
Final output
Once the displacements are complete, the gradients for all displacements are combined to give the full (or partial) Hessian. The Hessian is mass-weighted and diagonalized which results in the harmonic vibrational frequencies as eigenvalues and the normal modes as eigenvectors. Rotational and translational modes are projected out if the full Hessian is calculated.
An elemental normal mode composition factor analysis is automatically performed on the modes and thermochemistry based on the rigid-rotor-harmonic-oscillator (RRHO) approximation with a default temperature and pressure of 298 K and 1 atm.
Upon completion of the job, the full Hessian can be accessed in a few different ways:
It is stored in the hessian attribute of the ASH_Results object returned from the NumFreq function (see example below).
It is present in a file named "Hessian" in the "Numfreq_dir" (created by ASH). Can be read back into ASH using the read_hessian function.
It is present in an ORCA_formatted Hessian-file named "orcahessfile.hess". Can be read back into ASH using read_ORCA_Hessian
Additionally ASH creates a file "orcahessfile.hess_dummy.out" which is a dummy ORCA output file that contains the frequencies and normal modes. This file can be conveniently opened by some visualization programs such as Chemcraft and allows you to visualize the normal modes.
Examples:
Numerical frequencies in serial mode (QM-code parallelization instead used):
from ash import *
#the total number of CPU cores available to Ash (should match the job-script)
numcores=8
frag=Fragment(xyzfile="h2o.xyz", charge=0, mult=1)
#ORCA theory object, ORCA parallelization turned off by not providing numcores keyword
ORCAcalc = ORCATheory(orcasimpleinput="! r2SCAN-3c tightscf", numcores=numcores)
#Serial Numfreq job (default):
freqresult = NumFreq(fragment=frag, theory=ORCAcalc, npoint=2, runmode='serial')
print("freqresult:", freqresult)
#Print Hessian
print("Hessian:", freqresult.hessian)
The resulting object from a NumFreq calculation is an ASH_Results dataclass object. It contains the calculated frequencies, eigenvectors, normalmodes, list-of frequencies and a dictionary of thermochemical properties.
Numerical frequencies in parallel mode (QM-code parallelization turned off):
from ash import *
#the total number of CPU cores available to Ash (should match the job-script)
numcores=8
frag=Fragment(xyzfile="h2o.xyz", charge=0, mult=1)
#ORCA theory object, ORCA parallelization turned off by not providing numcores keyword
ORCAcalc = ORCATheory(orcasimpleinput="! r2SCAN-3c tightscf")
#Parallel mode: ASH will use the number of cores given to run same number of displacments simultaneously.
freqresult = NumFreq(fragment=frag, theory=ORCAcalc, npoint=2, runmode='parallel', numcores=numcores)
print("Vibrational frequencies (cm**-1) : ", freqresult.frequencies)
print("ZPVE (Eh) : ", freqresult.thermochemistry['ZPVE'])
print("Gibbs energy corrections (Eh) : ", freqresult.thermochemistry['Gcorr'])
Analytical frequencies
Some QM programs have analytical frequencies implemented and the ASH interface may support requesting the calculation of the analytical Hessian and reading Hessian back. Currently analytical frequencies are only supported in the QM codes: ORCATheory and CFourTheory
An analytical Hessian calculation is requested by the AnFreq function that takes fragment and theory as necessary arguments:
def AnFreq(fragment=None, theory=None, numcores=1, temp=298.15, pressure=1.0)
Example:
HF_frag=Fragment(xyzfile="hf.xyz")
ORCAcalc = ORCATheory(orcasimpleinput='BP86 def2-SVP def2/J tightscf', orcablocks="", numcores=1)
freqresult = AnFreq(theory=ORCAcalc, fragment=HF_frag)
print("Thermochem properties dict:", freqresult.thermochemistry)
print("Vibrational frequencies (cm**-1) : ", freqresult.frequencies)
thermochemistry corrections
Thermochemistry corrections are automatically calculated when either a Numfreq or Anfreq job is requested.
result_anfreq = AnFreq(theory=ORCAcalc, fragment=HF_frag)
result_numfreq = NumFreq(theory=ORCAcalc, fragment=HF_frag)
print("Thermochem property dict:", result_numfreq.thermochemistry)
print("ZPVE (Eh) : ", result_numfreq.thermochemistry['ZPVE'])
print("Gibbs energy corrections (Eh) : ", result_numfreq.thermochemistry['Gcorr'])
The return object from AnFreq or NumFreq contains a thermochemistry dictionary that contains the following information as dictionary keys. Note that the entropy terms (TS) are in energy units (Eh) as they have been multiplied by temperature T.
Key |
Property |
||
frequencies |
The harmonic vibrational frequencies (list). |
||
ZPVE |
Zero-point vibrational energy (harmonic) |
||
E_trans |
Translational energy at temp T. |
||
E_rot |
Translational energy at temp T. |
||
E_vib |
Vibrational energy at temp T. |
||
E_tot |
Total energy at temp T. |
||
TS_trans |
Translational entropy at temp T (in energy units). |
||
TS_rot |
Rotational entropy at temp T (in energy units) |
||
TS_vib |
Vibrational entropy at temp T (in energy units) |
||
TS_el |
Electronic entropy at temp T (in energy units) |
||
TS_tot |
Total entropy at temp T (in energy units) |
||
vibenergycorr |
Vibrational energy correction at temp T. |
||
Hcorr |
Total enthalpy correction at temp T. |
||
Gcorr |
Gibbs free energy correction at temp T. |
||
Alternatively, the thermochemcalc function can be called directly.
def thermochemcalc(vfreq,atoms,fragment, multiplicity, temp=298.18,pressure=1.0):
This function calculates the thermodynamic corrections from a list of available frequencies, list of atoms, ASH fragment object and spin multiplicity. The temperature (default: 298.15 K) and pressure (default: 1.0 atm) can be specified.
from ash import *
from ash.modules.module_freq import thermochemcalc
frag = Fragment(xyzfile="h2o.xyz", charge=0, mult=1)
#Manually defined frequencies for system
frequencies=[1600.1, 2300.2, 2400.3]
thermochemcalc(frequencies,frag.allatoms,frag, frag.mult, temp=298.18, pressure=1.0)
Calculating IR and Raman spectra
Calculation of IR and Raman spectra requires either the dipole derivatives or the polarizability derivatives along the normal modes. This means that to get IR/Raman intensitites from a numerical frequency calculation the dipole moment (IR) or polarizability (Raman) must be calculated for each displacement by the QM-code.
IR/Raman spectra can be calculated for any QM-level of theory for which analytical gradients (first derivatives) are available and if dipole moments/polarizabilities are available in the code or supported by ASH interface (see below). QM/MM theories within ASH are also supported.
IR intensities:
In the ASH interfaces to ORCA, pySCF, xTB, MRCC, CFour, Block and Dice, the dipole moments are automatically available and the NumFreq module will in this case get the dipole moment from each calculation and automatically calculate the dipole derivatives along the normal modes and provide the final IR intensity for each normal mode. The IR intensities are stored in the ASH_Results object returned from the NumFreq function.
Example: IR intensities using ORCA
from ash import *
frag=Fragment(databasefile="h2o.xyz", charge=0, mult=1)
#ORCA theory
ORCAcalc = ORCATheory(orcasimpleinput="! r2SCAN-3c tightscf", numcores=4)
#Serial Numfreq job
result = NumFreq(fragment=frag, theory=ORCAcalc, npoint=2, runmode='serial')
print("Vibrational frequencies (cm**-1) : ", result.frequencies)
print("IR intensities (km/mol): ", result.IR_intensities)
Raman activities:
Raman activities are a bit more complicated. Analytic polarizabilities are not always available in the QM-code and they are much more expensive. Polarizabilities are available in the ASH interface to ORCA, pySCF and CFour. To get the Raman activities one must i) tell the QM-code to calculate polarizabilities and ii) tell the NumFreq module that one wants Raman (Raman=True).
Example: Raman activities using ORCA
from ash import *
frag=Fragment(databasefile="h2o.xyz", charge=0, mult=1)
# ORCA Theory requesting polarizability calculation
blocks="""
%elprop
polar 1
end
"""
ORCAcalc = ORCATheory(orcasimpleinput="! r2SCAN-3c tightscf", orcablocks=blocks, numcores=4)
#NumFreq
result = NumFreq(fragment=frag, theory=ORCAcalc, npoint=2, runmode='serial', Raman=True)
print("Vibrational frequencies (cm**-1) : ", result.frequencies)
print("Raman activities (Å^4/amu): ", result.Raman_activities)
Plot IR/Raman spectra:
The vibrational frequencies, IR intensities and Raman activitites can be found in the ASH output:
----------------------------------------
VIBRATIONAL FREQUENCY SUMMARY
----------------------------------------
Note: imaginary modes shown as negative
Mode Freq(cm**-1) IR Int.(km/mol) Raman Act.(Å^4/amu)
0 0.0000 0.0000 0.0000 (TR mode)
1 0.0000 0.0000 0.0000 (TR mode)
2 0.0000 0.0000 0.0000 (TR mode)
3 0.0000 0.0000 0.0000 (TR mode)
4 0.0000 0.0000 0.0000 (TR mode)
5 0.0000 0.0000 0.0000 (TR mode)
6 1608.3146 81.7550 1.3155
7 3851.9971 4.3099 74.7428
8 3993.9783 63.5039 22.0644
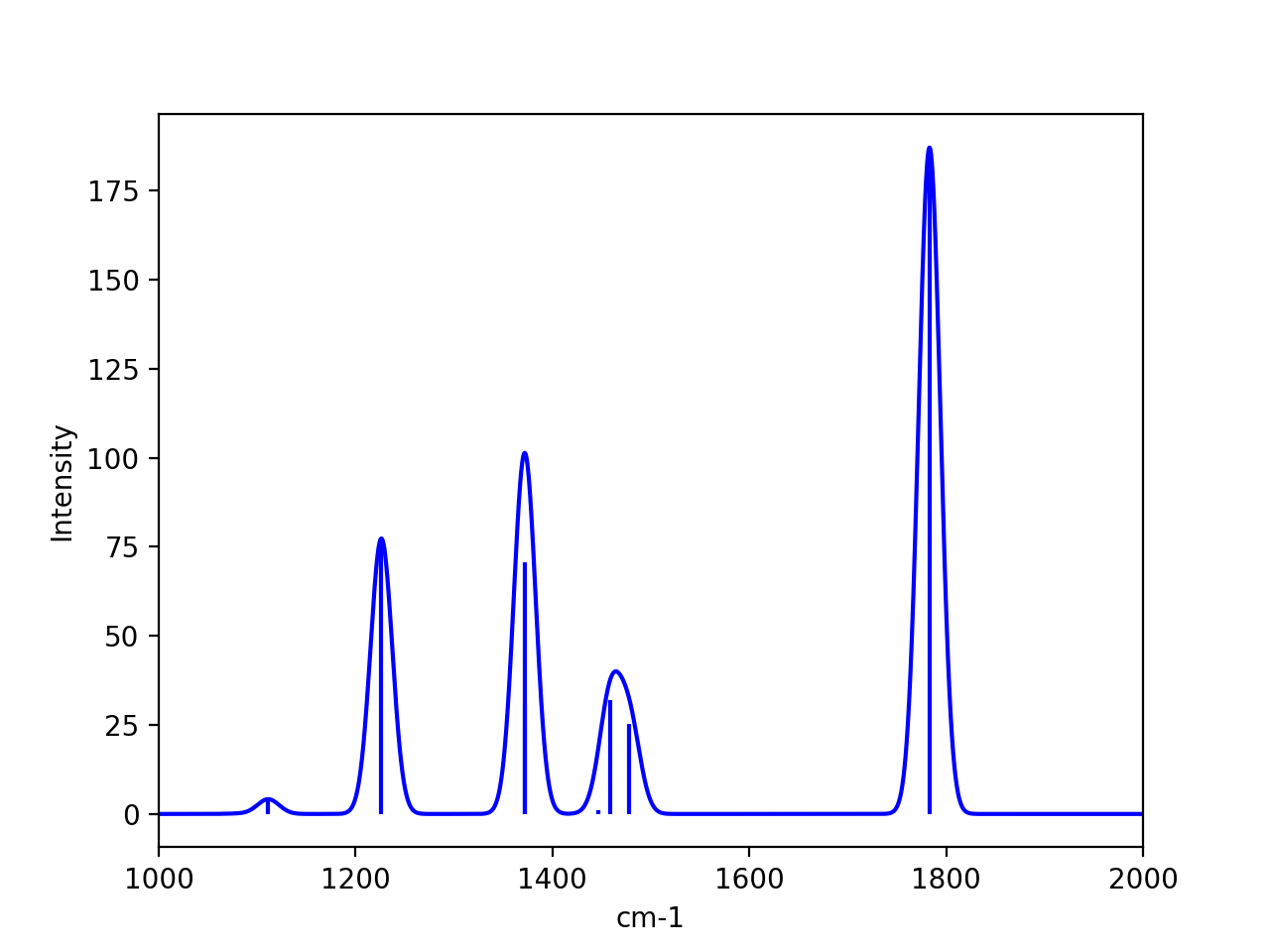
and can also be found in the ASH Results object. If one wants to plot a broadened IR/Raman spectra one can use plot_Spectrum function (see Plotting), providing the frequencies and intensities from the Results object as x- and y-values.
from ash import *
frag=Fragment(databasefile="acetone.xyz", charge=0, mult=1)
#ORCA theory object
ORCAcalc = ORCATheory(orcasimpleinput="! r2SCAN-3c", numcores=4)
#Optimization
Optimizer(fragment=frag, theory=ORCAcalc)
#Frequencies
result = NumFreq(fragment=frag, theory=ORCAcalc, npoint=2, runmode='serial')
print("Vibrational frequencies (cm**-1) : ", result.frequencies)
print("IR intensities (km/mol): ", result.IR_intensities)
#Plotting broadened IR spectrum (11 cm-1 broadening, 5000 points) for 1000-2000 cm-1 window.
plot_Spectrum(xvalues=result.frequencies, yvalues=result.IR_intensities, plotname='IR_spectrum', range=[1000,2000], unit='cm-1',
broadening=11, points=5000, imageformat='png', dpi=200)
Modifying masses
If specific masses (isotopes) are wanted when calculating vibrational frequencies, then this can be done by providing a list of masses (hessatoms_masses keyword) to the NumFreq function. If you have already performed a NumFreq calculation and want to see the effect of mass-change/isotope substitution then this can be performed by reading in the ASH_Results object from a file, grabbing the data needed, changing masses and then re-diagonalize a Hessian with new masses.
from ash import *
frag = Fragment(databasefile="h2o.xyz")
#Assuming previous calculation run like the commented-out lines below
#theory = PySCFTheory(scf_type="RHF",basis="def2-SVP", printlevel=1)
# NumFreq
#result = NumFreq(theory=theory,fragment=frag, Raman=True)
#Writing Result object to disk (nowadays this is default)
#result.write_to_disk(filename="ASH.result")
#Read Result-file from disk (shown as this can be used to avoid repeating the above Numfreq calculation)
result = read_results_from_file(filename="ASH_NumFreq.result")
print(result)
#Print the previous masses (for comparison)
print("Current Hessian masses:", result.freq_masses)
#Defining new masses
hessmasses= [18.0, 1.0, 1.0]
#Grabbing previous data from ASH-Results object (Polarizability data can be skipped if not available)
hessian=result.hessian
hesselems=result.freq_elems
hessatoms=result.freq_atoms
hesscoords=result.freq_coords
TRmodenum=result.freq_TRmodenum
projection=result.freq_projection
scaling_factor=result.freq_scaling_factor
dipole_derivs=result.freq_dipole_derivs
polarizability_derivs=result.freq_polarizability_derivs
polarizability_derivs = [np.array(i) for i in polarizability_derivs]
Raman=result.freq_Raman
#Diagonalize Hessian
frequencies, nmodes, evectors, mode_order = ash.modules.module_freq.diagonalizeHessian(hesscoords,hessian,hessmasses,hesselems,TRmodenum=TRmodenum,projection=projection)
print("Diagonalization of frequencies complete")
print("Now scaling frequencies by scaling factor:", scaling_factor)
frequencies = scaling_factor * np.array(frequencies)
# IR intensities if dipoles available
IR_intens_values = ash.modules.module_freq.calc_IR_Intensities(hessmasses,evectors,dipole_derivs)
# Raman activities if polarizabilities available
Raman_activities, depolarization_ratios = ash.modules.module_freq.calc_Raman_activities(hessmasses,evectors,polarizability_derivs)
# Print out Freq output.
ash.modules.module_freq.printfreqs(frequencies,len(hessatoms),TRmodenum=TRmodenum, intensities=IR_intens_values,
Raman_activities=Raman_activities)
# Print thermochemistry for new vibrational frequencies
thermodict = ash.modules.module_freq.thermochemcalc(frequencies,hessatoms, frag, frag.mult)