Metalloprotein tutorial II: Ferredoxin
If you have not gone through the rubredoxin tutorial (Metalloprotein tutorial I: Rubredoxin ) before, then please do so before. Here we encounter a slightly more complex metalloprotein example, ferredoxin. This protein setup require slightly more work due to errors arising parsing the more complicated PDB-file with a slightly more complicated metal cluster.
Ferredoxin contains 2 copies of the [2Fe-2S] cofactor bound to 4 deprotonated cysteines. Additionally, the X-ray structure contains additional molecules: SCN- and SO42- anions and a benzamidine molecule.
The files for this tutorial can be found in the ASH source code directory under $ASHDIR/examples/OpenMM_Modeller-setups/ferredoxin or Github-repository
1. OpenMM_Modeller PDB parsing errors
If we download the 6lk1.pdb file and read into OpenMM_Modeller:
from ash import *
#Original raw PDB-file (no hydrogens, nosolvent)
pdbfile="6lk1.pdb"
# Setting up system via Modeller
OpenMM_Modeller(pdbfile=pdbfile,forcefield="CHARMM36")
1a. Multiple occupancy problems
we will get this error:
Found residues in PDB-file that have alternate location labels i.e. multiple occupancies:
Chain A:
ILE23
GLU28
...
Chain B:
ASP20
ILE23
...
These residues should be manually inspected and fixed in the PDB-file before continuing.
You should delete either the labelled A or B location of the residue-atom/atoms and then remove the A/B label from column 17 in the file
Alternatively, you can choose use_higher_occupancy=True keyword in OpenMM_Modeller and ASH will keep the higher occupied form and go on
Make sure that there is always an A or B form present.
Exiting.
ASH exiting with code: 1
This means that the PDB-file contains multiple occupancies for some atoms of some residues in the PDB-file. It is best to now visually inspect these residues in the PDB, using e.g. VMD, before continuing. Depending on the importance of these residues you may choose to keep either form A or B of these residues. As an example for residue ILE23 in chain A in the PDB-file we have:
ATOM 168 N ILE A 23 4.324 3.322 14.973 1.00 5.48 N
ANISOU 168 N ILE A 23 764 686 507 94 -97 -89 N
ATOM 169 CA AILE A 23 3.083 3.972 15.441 0.72 5.57 C
ANISOU 169 CA AILE A 23 714 705 572 141 -15 -72 C
ATOM 170 CA BILE A 23 3.078 3.888 15.468 0.28 5.59 C
ANISOU 170 CA BILE A 23 805 655 539 144 20 -3 C
ATOM 171 C ILE A 23 3.137 4.183 16.956 1.00 5.62 C
ANISOU 171 C ILE A 23 774 668 567 89 -73 -83 C
ATOM 172 O ILE A 23 2.785 5.260 17.422 1.00 6.14 O
ANISOU 172 O ILE A 23 946 662 584 162 -104 -74 O
ATOM 173 CB AILE A 23 1.848 3.204 14.948 0.72 7.07 C
ANISOU 173 CB AILE A 23 874 1044 607 -20 -54 -250 C
ATOM 174 CB BILE A 23 1.888 2.931 15.191 0.28 5.35 C
ANISOU 174 CB BILE A 23 650 656 605 357 -287 -91 C
ATOM 175 CG1AILE A 23 1.736 3.459 13.417 0.72 9.07 C
ANISOU 175 CG1AILE A 23 1360 1209 672 245 -314 -256 C
ATOM 176 CG1BILE A 23 1.631 2.627 13.721 0.28 6.19 C
ANISOU 176 CG1BILE A 23 584 1013 616 458 -190 -102 C
ATOM 177 CG2AILE A 23 0.549 3.592 15.635 0.72 11.02 C
ANISOU 177 CG2AILE A 23 711 2093 1133 141 -68 -310 C
ATOM 178 CG2BILE A 23 0.628 3.478 15.877 0.28 4.77 C
ANISOU 178 CG2BILE A 23 586 572 545 411 -433 -162 C
ATOM 179 CD1AILE A 23 0.876 2.420 12.788 0.72 13.20 C
ANISOU 179 CD1AILE A 23 1175 2543 1000 -202 -285 -837 C
ATOM 180 CD1BILE A 23 1.064 3.763 12.898 0.28 5.93 C
ANISOU 180 CD1BILE A 23 1050 781 287 36 33 29 C
where the lines indicate that there is multiple occupancy of sites A and B for some atoms with occupancy of 0.72 and 0.28 respectively. If we visualize this residue in the PDB-file using VMD we see that the multiple occupancy corresponds to slightly different conformers of the ILE sidechain. For a flexible residue like ILE this is not surprising as this would most likely arise due to dynamic disorder. and we can simply choose either conformer A or B and proceed.
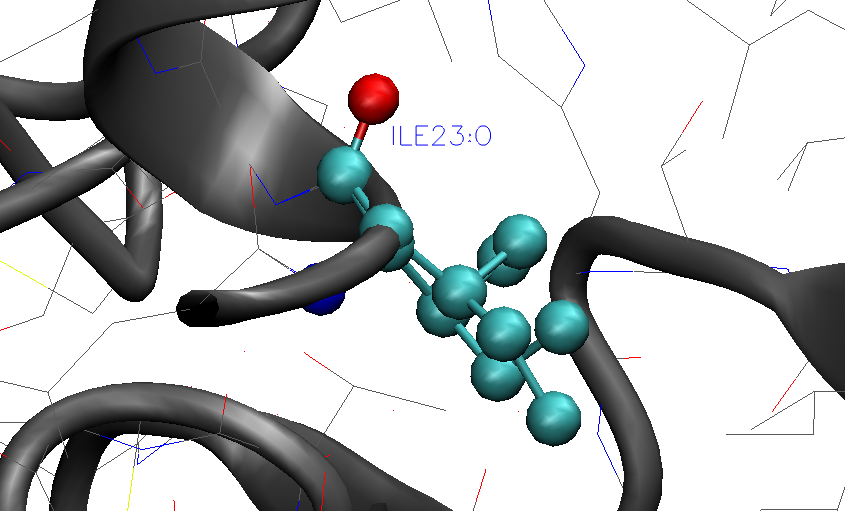
For this example we would could delete e.g. the lines for conformer B (lower occupancy) and remove the 'A' characters in front of 'ILE' for the lines corresponding to site A.
If, after visual inspection of all residues we are confident that the multiple occupancy present is not a significant issue (e.g. simple dynamic disorder) and that it should be fine to simply keep the higher occupancy conformer then we can use the keyword option, use_higher_occupancy=True, to proceed. This will simply keep the higher occupancy version of each atom that has multiple occupancy and proceed:
from ash import *
#Original raw PDB-file (no hydrogens, nosolvent)
pdbfile="6lk1.pdb"
# Setting up system via Modeller
OpenMM_Modeller(pdbfile=pdbfile,forcefield="CHARMM36", use_higher_occupancy=True)
1b. Missing forcefield parameters for FeS cluster
The next problem encountered occurs as OpenMM tries to apply the chosen forcefield to the system topology of the PDB-file.
Error: OpenMM modeller.addHydrogens signalled a ValueError
This is a common error and suggests a problem in PDB-file or missing residue information in the forcefield.
Non-standard inorganic/organic residues require providing an additional XML-file via extraxmlfile= option
Note that C-terminii require the dangling O-atom to be named OXT
Read the ASH documentation or the OpenMM documentation on dealing with this problem.
Full error message from OpenMM:
No template found for residue 190 (FES). This might mean your input topology is missing some atoms or bonds,
or possibly that you are using the wrong force field.
This arises simply because the FES residue in the PDB-file, i.e. the [2Fe-2S] cluster is not present in the CHARMM36 forcefield. In the PDB-file the residue is defined like this:
HETATM 1457 FE1 FES A 101 5.508 -6.531 22.895 1.00 7.55 FE
HETATM 1458 FE2 FES A 101 4.734 -4.618 21.086 1.00 6.50 FE
HETATM 1459 S1 FES A 101 5.095 -4.352 23.250 1.00 7.17 S
HETATM 1460 S2 FES A 101 4.997 -6.752 20.748 1.00 7.46 S
If we tell OpenMM_Modeller about the extra residue:
from ash import *
#Original raw PDB-file (no hydrogens, nosolvent)
pdbfile="6lk1.pdb"
#XML-file to deal with FeS cluster
extraxmlfile="specialresidue.xml"
# Setting up system via Modeller
OpenMM_Modeller(pdbfile=pdbfile,forcefield="CHARMM36", use_higher_occupancy=True,
extraxmlfile="specialresidue.xml")
where specialresidue.xml contains:
<ForceField>
<AtomTypes>
<Type name="FEX" class="Fe" element="Fe" mass="55.84700"/>
<Type name="SXM" class="S" element="S" mass="32.065"/>
</AtomTypes>
<Residues>
<Residue name="FES">
<Atom name="FE1" type="FEX"/>
<Atom name="FE2" type="FEX"/>
<Atom name="S1" type="SXM"/>
<Atom name="S2" type="SXM"/>
</Residue>
</Residues>
<NonbondedForce coulomb14scale="1.0" lj14scale="1.0">
<Atom type="FEX" charge="0.0" sigma="1.0" epsilon="0.0"/>
<Atom type="SXM" charge="0.0" sigma="1.0" epsilon="0.0"/>
</NonbondedForce>
<LennardJonesForce lj14scale="1.0">
<Atom type="FEX" sigma="0.194215920554594" epsilon="1.046"/>
<Atom type="SXM" sigma="0.35190499366543404" epsilon="1.58992"/>
</LennardJonesForce>
</ForceField>
Here we have chosen, like for rubredoxin, to define LJ parameters (sigma and epsilon) for the Fe ion, using available parameters in the CHARMM forcefield for Zn. Charges for Fe and S in the [2Fe-2S] cluster are for now set to 0.0 but will be changed to appropriate values later.
Warning
For OpenMM to correctly map the information from the specialresidue.xml onto the PDB-file topology, it is important that the PDB-file contains an element definition (column 77-78) for each element of the special residue.
Running this script we may get instead another error (OpenMM version specific):
ValueError: No template found for residue 191 (FES). The set of atoms matches FES, but the bonds are different.
Perhaps the chain is missing a terminal group?
This error messages suggest that OpenMM Modeller recognizes our residue definition for FES but is confused about bonding. This arises due to the presence of bonding information in the bottom of the PDB-file in the form of CONE lines (each line indicates which atom indices should be considered to have a bond between them):
CONECT 299 1457
CONECT 338 1457
CONECT 356 1458
CONECT 575 1458
CONECT 1033 1523
CONECT 1064 1523
CONECT 1082 1524
CONECT 1298 1524
CONECT 1457 299 338 1459 1460
CONECT 1458 356 575 1459 1460
CONECT 1459 1457 1458
While we could add bonding information to specialresidue.xml and try to match the connectivity in the PDB-file an easier solution is to remove the connectivity information by creating a modified version of the PDB-file. This should probably always be an acceptable solution since we will constrain our metal-cluster residue anyway, as we don't have forcefield parameters available. Thus we make a modified version, called 6lk1-mod.pdb, that does not contain the CONE lines and we also remove most header lines of the PDB-file (all lines before ATOM/HETATM section begins).
Next we run our script again (now using 6lk1-mod.pdb as input PDB file):
from ash import *
#Original raw PDB-file (no hydrogens, nosolvent)
pdbfile="6lk1-mod.pdb"
#XML-file to deal with cofactor
extraxmlfile="specialresidue.xml"
# Setting up system via Modeller
OpenMM_Modeller(pdbfile=pdbfile,forcefield="CHARMM36", use_higher_occupancy=True,
extraxmlfile="specialresidue.xml")
1c. Unrecognized ligands in PDB-file
When we run this script we instead get a different error message:
ValueError: No template found for residue 192 (BEN). The set of atoms is similar to INDA, but it is missing 6 hydrogen atoms.
This message refers to the fact that the PDB-file contains BEN residues (benzamidine) that are neither present in the CHARMM36 protein forcefield or in our specialresidue.xml file. As benzamidine is simply a crystallized molecule from the buffer solution and has little do with the protein, we here make the choice to simply remove the BEN residues from 6lk1-mod.pdb.
We then get similar messages associated with missing residue definitions for SCN and SO4
ValueError: No template found for residue 191 (SCN). The set of atoms is similar to THAZ, but it is missing 5 atoms.
ValueError: No template found for residue 191 (SO4). The set of atoms is similar to MSO4, but it is missing 4 atoms.
and again we make the choice to remove these crystallized contaminants from 6lk1-mod.pdb. Once we have done this, OpenMM_Modeller proceeds without problems but this does not mean of course that the system is correctly set up.
2. OpenMM residue variants: protonation states
As previously occurred for rubredoxin, OpenMM Modeller protonates the cysteine residues that are coordinated to the Fe ions.
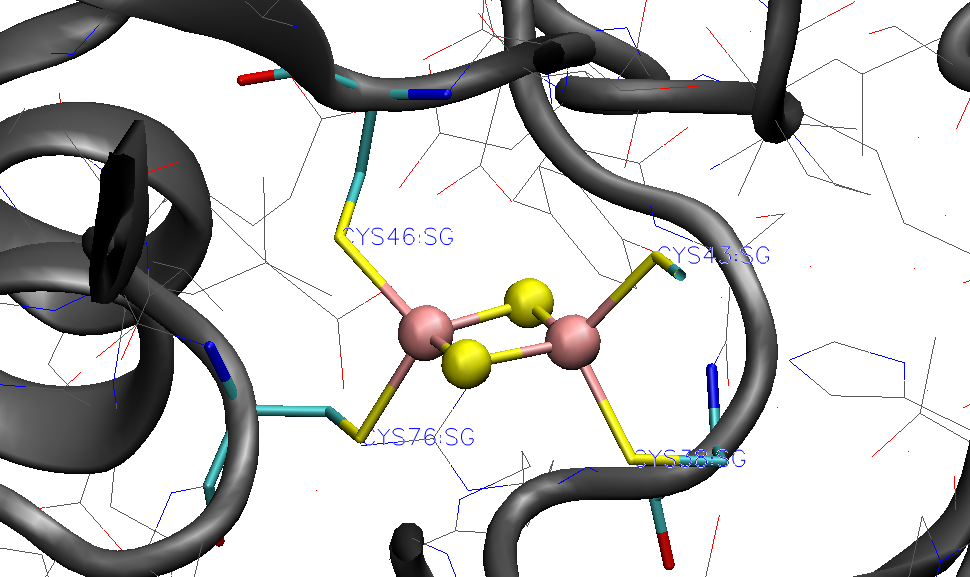
Since we want to avoid this, we again define a dictionary with information about abnormal residues and pass this on to OpenMM_Modeller. Since the protein contains two protein chains (named 'A' and 'B' in the PDB-file) with the [2Fe-2S] cofactor coordinated to 4 cysteines in each chain, we need to define all of these cysteines to be deprotonated ('CYX' label) in each chain.
from ash import *
#Original raw PDB-file (no hydrogens, nosolvent)
pdbfile="6lk1-mod.pdb"
#XML-file to deal with FeS cluster
extraxmlfile="specialresidue.xml"
#Defining deptonated cysteine residues
residue_variants={'A':{38:'CYX',43:'CYX',46:'CYX',76:'CYX'},'B':{38:'CYX',43:'CYX',46:'CYX',76:'CYX'}}
# Setting up system via Modeller
OpenMM_Modeller(pdbfile=pdbfile,forcefield="CHARMM36", use_higher_occupancy=True,
extraxmlfile="specialresidue.xml", residue_variants=residue_variants)
The printed table shows what Cys residues we selected to deprotonate:
User defined residue variants per chain:
Chain A : {38: 'CYX', 43: 'CYX', 46: 'CYX', 76: 'CYX'}
Chain B : {38: 'CYX', 43: 'CYX', 46: 'CYX', 76: 'CYX'}
MODELLER TOPOLOGY - RESIDUES TABLE
ASH-resid Resname Chain-index Chain-name ResID-in-chain User-modification
----------------------------------------------------------------------------------------------------
0 ALA 0 A 1
1 TYR 0 A 2
2 LYS 0 A 3
...
37 CYS 0 A 38 -- This residue will be changed to: CYX --
38 ARG 0 A 39
39 ALA 0 A 40
40 GLY 0 A 41
41 ALA 0 A 42
42 CYS 0 A 43 -- This residue will be changed to: CYX --
43 SER 0 A 44
44 SER 0 A 45
45 CYS 0 A 46 -- This residue will be changed to: CYX --
...
75 CYS 0 A 76 -- This residue will be changed to: CYX --
In this tutorial we only change the protonation states of the Cys residues but a more careful study should visually inspect the protonation states of all titratable residues in the protein: HIS, GLU, ASP, ARG, LYS. When these residues are on the surface of the protein they can usually be assumed to be in their regular free-form protonation states (e.g deprotonated (ASP,GLU), protonated (LYS,ARG), singly-protonated (HIS)). However, when these residues are buried in a protein the local pH may be quite different and residues may be in a different protonation state.
Programs like PROPKA may be used to help in determining protonation states of residues: https://github.com/jensengroup/propka https://server.poissonboltzmann.org
When the X-ray structure of the protein is of sufficient quality the hydrogen bonding network may more clearly reveal whether a residue should be protonated or not. Special attention should be paid to the protonation state of HIS residue as it can be both doubly and singly protonated and when singly protonated, it can protonated on the delta-nitrogen or the epsilon-nitrogen of the imidazole ring. The hydrogen bonding network in the X-ray structure can often be used to make this assignment. In rare cases, note that the atoms of the imidazole ring of HIS may have been mis-assigned due to the similar electron densities around carbon and nitrogen. This can be spotted if you see e.g. a carbon atom of the imidazole ring in hydrogen-bonding distance to another heavy atom. To fix such a problem requires modifying the coordinates in the PDB-file.
3. A more realistic nonbonded model for the [2Fe-2S]
While a pragmatic solution to dealing with simple inorganic residues like the [2Fe-2S] cluster is to simply create dummy forcefield parameters in the specialresidue.xml file to begitn with, this will likely lead to artifacts in an MD simulation. If the charges of the Fe and S atoms in [2Fe-2S] are zero, then this means no electrostatic interaction is present between these atoms and the rest of the protein+solvent during a classical MD simulation. Furthermore, if epsilon or sigma parameters are 0.0 then no repulsion (or attractive dispersion) forces are present between [2Fe-2S] and other atoms, meaning that other atoms could start to occupy the same space as the [2Fe-2S] cluster.
Note
In electrostatically embedded QM/MM the metal cluster will most often be in the QM-region and any atom charges defined for the cluster will not be used. Note, however, that the LJ interactions between QM and MM atoms are calculated and the LJ parameters may be important.
Thus a more realistic scenario is to come up with a proper nonbonded model for the [2Fe-2S] cluster: i.e. atomic charges and Lennard-Jones parameters. There are 3 main choices here:
Search the literature for a study using nonbonded MM parameters for the same/similar residue. Ideally with the same protein forcefield.
Derive the parameters using similar residues already present in the forcefield.
Derive the parameters from a DFT calculation and a population analysis.
Option 3 is the more general solution but as only nonbonded parameters are derived, this furthermore requires the residues to be frozen/constrained/restrained during a classical MD simulation.
ASH features a convenient way of automatically creating a valid XML-file with appropriate nonbonded parameters for an inorganic residue. More info here
from ash import *
frag=Fragment(xyzfile="Fe2S2.xyz", charge=2, mult=1)
#Script to get nonbonded model parameters for a ligand
orcatheory=ORCATheory(orcasimpleinput="! UKS r2scan ZORA ZORA-def2-TZVP tightscf CPCM", numcores=1,
brokensym=True, HSmult=11, atomstoflip=[0])
write_nonbonded_FF_for_ligand(fragment=frag, theory=orcatheory, resname="test",
coulomb14scale=1.0, lj14scale=1.0, charmm=True, charge_model="CM5_ORCA", LJ_model="UFF")
Here we read the inorganic residue coordinates from an XYZ-file into an ASH fragment, define an appropriate level of theory, and then call the ASH function write_nonbonded_FF_for_ligand giving the fragment and theory objects as input. We choose the coulomb14scale and lj14scale parameters to be consistent with the CHARMM forcefield (1.0) and tell the function to create an XML-file in CHARMM forcefield format (separate <NonbondedForce and <LennardJonesForce blocks in the XML-file). Finally we tell the function to derive CM5 atomic charges (scaled Hirshfeld charges) and to use element-specific UFF Lennard-Jones parameters for the atoms of the residue. This will create a file, here called test.xml that contains a forcefield definition for the residue that could be used instead of the dummy file previously used.
<ForceField>
<AtomTypes>
<Type name="FeX0" class="FeX0" element="Fe" mass="55.845"/>
<Type name="FeX1" class="FeX1" element="Fe" mass="55.845"/>
<Type name="SX2" class="SX2" element="S" mass="32.065"/>
<Type name="SX3" class="SX3" element="S" mass="32.065"/>
</AtomTypes>
<Residues>
<Residue name="MCMtest">
<Atom name="FeY0" type="FeX0"/>
<Atom name="FeY1" type="FeX1"/>
<Atom name="SY2" type="SX2"/>
<Atom name="SY3" type="SX3"/>
</Residue>
</Residues>
<NonbondedForce coulomb14scale="1.0" lj14scale="1.0">
<Atom type="FeX0" charge="1.0271058194510412" sigma="0.0" epsilon="0.0"/>
<Atom type="FeX1" charge="1.007310147382089" sigma="0.0" epsilon="0.0"/>
<Atom type="SX2" charge="-0.013198936151894144" sigma="0.0" epsilon="0.0"/>
<Atom type="SX3" charge="-0.02122203068123596" sigma="0.0" epsilon="0.0"/>
</NonbondedForce>
<LennardJonesForce lj14scale="1.0">
<Atom type="FeX0" sigma="0.2594297067224668" epsilon="0.054392"/>
<Atom type="FeX1" sigma="0.2594297067224668" epsilon="0.054392"/>
<Atom type="SX2" sigma="0.3594776327696269" epsilon="1.146416"/>
<Atom type="SX3" sigma="0.3594776327696269" epsilon="1.146416"/>
</LennardJonesForce>
</ForceField>
There are several caveats associated with this approach:
We derive the parameters here for a bare [2Fe-2S] cluster, ignoring the fact that this cluster is bound to the protein via 4 Cys residues. An alternative approach would use some kind of capping groups to give a more realistic ligand-field for the [2Fe-2S] cluster, e.g. 4 SH- or SMe groups. This would, however, require deleting the capping atoms afterwards and the total charge of the bare [2Fe-2S] cluster would also have to be adjusted as it must add up to full charge of the cluster (some charge-transfer always takes place between residues in a QM calculation).
The theory level (here ZORA-r2SCAN with a TZ basis set) and population analysis (here CM5) will affect the values of the calculated charges and is here chosen as a somewhat realistic level to describe iron-sulfur clusters. However, the atomic charges defined like this are not necessarily compatible with how charges are defined in the protein forcefield, here CHARMM. In CHARMM, atomic charges for residues are typically defined at the HF/6-31G* level level of theory which is unrealistic for transition metal complexes. A better way to deal with this problem might involve scaling the atomic charges so that the magnitude of the charges are more in line with the charges of protein residues. It is unclear what the best way to do this would be.
The write_nonbonded_FF_for_ligand function does not recognize the symmetry of the cluster and will create an atomtype for each atom in the cluster. This might be undesirable and the user may choose to instead combine atomtypes, e.g. a common atomtype for the Fe and S ions, respectively.
The UFF parameters are not ideal as they are element-specific and do not take into account different oxidation states of Fe and S.
4. Classical MD simulation
TO BE DONE...