Tutorial: QM/MM boundary
The definition of the QM-region (a list of atom indices) is important when modelling proteins by QM/MM as the QM/MM boundary will be defined accordingly. ASH will not allow any type of boundary to be defined, only certain types of cuts are allowed.
Most QM/MM programs (including ASH) do not allow a situation where 2 linkatoms get added to the same dangling QM-atom (ASH will complain if you try to do this). Thus one must always avoid such a boundary.
The standard linkatom strategy in QM/MM is not perfect but it works extremely well as long as the following conditions are met:
You make a QM-MM boundary by cutting through a single plain bond (no double or triple cuts)
The bond in question is ideally a plain carbon-carbon bond (no heteronuclear bonds, e.g. C-O or C-N)
The C-C bond is as nonpolar as possible (can not always be fulfilled).
You don‘t cut throuch a charge-group of the MM forcefield.
ASH will exit if the QM-region definition leads to a situation where 2 linkatoms would be added to the same dangling QM-atom. ASH will also exit if the QM-MM definition leads to a heteronuclear bond being cut through (e.g. C-O or C-N) as this is rarely desired. However, this behaviour can be overridden via the use of the keyword option unusualboundary=True .
Since knowledge of CHARMM charge groups in amino acid residues can influence our QM-region choice we show here the CHARMM residue definitions for deprotonated cysteine (named CYM by CHARMM) and valine (VAL). This can be found in CHARMM top_all36_prot.rtf files.
RESI CYM -1.00 ! Anionic Cysteine
ATOM N NH1 -0.47 ! |
ATOM HN H 0.31 ! HN-N
ATOM CA CT1 0.07 ! | HB1
ATOM HA HB1 0.09 ! | | -
GROUP ! HA-CA--CB--SG (thiolate)
ATOM CB CS -0.38 ! | |
ATOM HB1 HA2 0.09 ! | HB2
ATOM HB2 HA2 0.09 ! O=C
ATOM SG SS -0.80 ! |
GROUP
ATOM C C 0.51
ATOM O O -0.51
RESI VAL 0.00
GROUP
ATOM N NH1 -0.47 ! | HG11 HG12
ATOM HN H 0.31 ! HN-N | /
ATOM CA CT1 0.07 ! | CG1--HG13
ATOM HA HB1 0.09 ! | /
GROUP ! HA-CA--CB-HB
ATOM CB CT1 -0.09 ! | \
ATOM HB HA1 0.09 ! | CG2--HG21
GROUP ! O=C / \
ATOM CG1 CT3 -0.27 ! | HG21 HG22
ATOM HG11 HA3 0.09
ATOM HG12 HA3 0.09
ATOM HG13 HA3 0.09
GROUP
ATOM CG2 CT3 -0.27
ATOM HG21 HA3 0.09
ATOM HG22 HA3 0.09
ATOM HG23 HA3 0.09
GROUP
ATOM C C 0.51
ATOM O O -0.51
RESI THR 0.00
GROUP
ATOM N NH1 -0.47 ! |
ATOM HN H 0.31 ! HN-N
ATOM CA CT1 0.07 ! | OG1--HG1
ATOM HA HB1 0.09 ! | /
GROUP ! HA-CA--CB-HB
ATOM CB CT1 0.14 ! | \
ATOM HB HA1 0.09 ! | CG2--HG21
ATOM OG1 OH1 -0.66 ! O=C / \
ATOM HG1 H 0.43 ! | HG21 HG22
GROUP
ATOM CG2 CT3 -0.27
ATOM HG21 HA3 0.09
ATOM HG22 HA3 0.09
ATOM HG23 HA3 0.09
GROUP
ATOM C C 0.51
ATOM O O -0.51
Defining different QM-region for an Fe-cysteine protein (rubredoxin).
Here we will use a QM/MM model of rubredoxin as an example. See Metalloprotein tutorial I: Rubredoxin on how to set up the system using the CHARMM36 forcefield. You can find the files for this MM-setup in the Github-repository . For ASH scripts related to the QM-region definitions in this tutorial see directory named QM-MM-boundary-setup
In rubredoxin the Fe ion is bound to 4 cysteine residues. It is always best to try to start as simple as possible and figure out what the minimal QM-region is for a decent description of the system. Note that visualizing the PDB-file of the full system (containing all atoms) in a visualization program like VMD is probably the best way to figure out what a good QM-region should look like and to get the atom indices involved (VMD like ASH will count atom indices from 0). Visualizing the PDB-file is better than an XYZ-file because VMD will then show atomnames and residuenames and resids when you select individual atoms.
Option 1: Only Fe and S-atom in the QM-region (BAD)
We can first try to define the QM-region to only include the Fe atom (no. 755) and the 4 S atoms of the 4 bound cysteines (96,136,567,607).
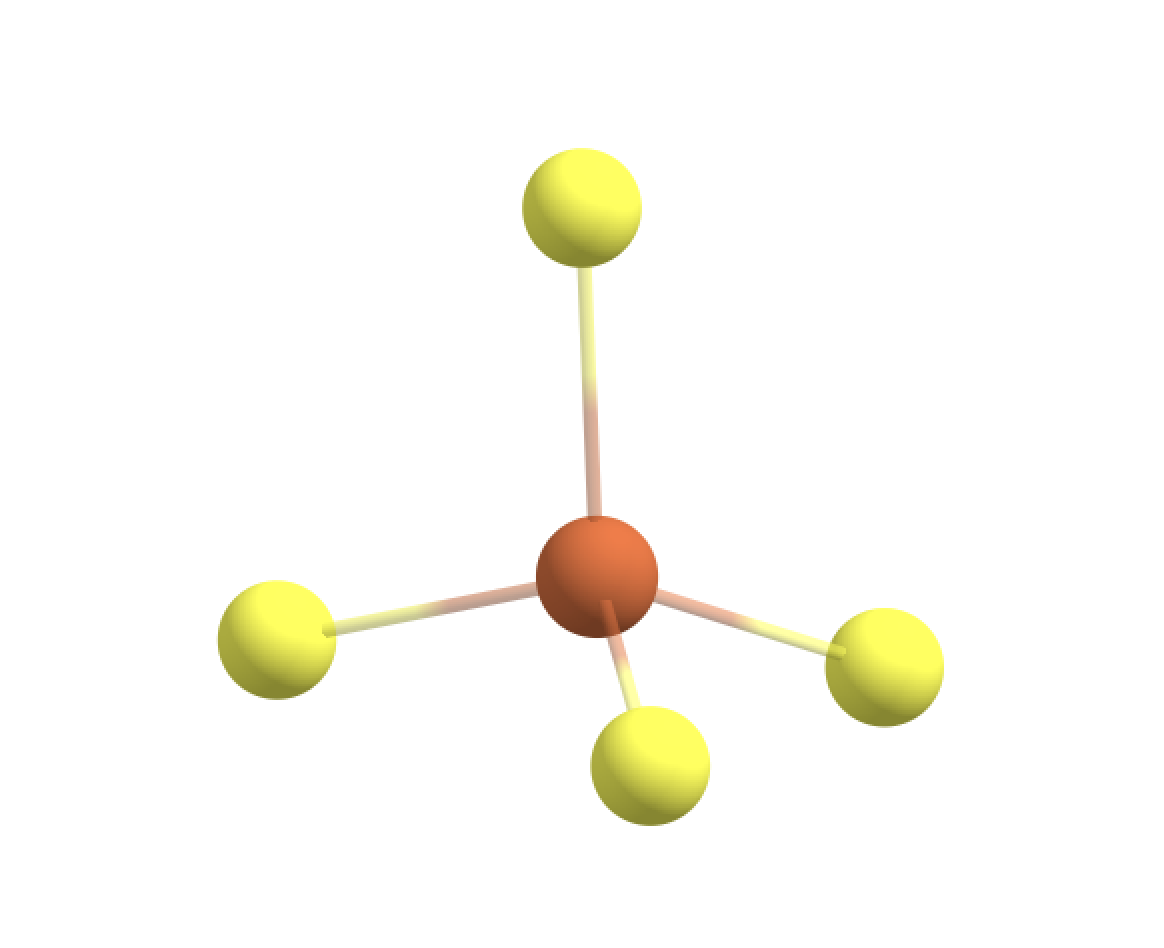
#Note that ASH counts from 0 (not 1).
qmatoms=[96,136,567,607,755]
#Note: unusualboundary=True keyword is ne
qmmm = QMMMTheory(qm_theory=orca, mm_theory=omm, fragment=fragment, qm_charge=-1, qm_mult=6,
qmatoms=qmatoms, unusual_boundary=True)
ASH would print the following in the output:
QM-region coordinates (before linkatoms):
96 S 28.35000000 36.95500000 29.37100000
136 S 32.02800000 37.58300000 29.84000000
567 S 30.30900000 34.16000000 30.68100000
607 S 30.11200000 37.06500000 32.95600000
755 Fe 30.20200000 36.48400000 30.72700000
Determining QM-MM boundary
Parameters determing connectivity:
Scaling factor: 1.0
Tolerance: 0.4
QM atoms: [96, 136, 567, 607, 755]
QM atoms to be excluded from boundary creation (excludeboundaryatomlist): []
Warning: QM-MM boundary is not the ideal C-C scenario:
QM-MM boundary: S(96) - C(93)
Make sure you know what you are doing (also note that ASH counts atoms from 0 not 1). Exiting.
To override exit, add: unusualboundary=True to QMMMTheory object
ASH exiting with code: 1
ASH here exited actually, and this is because it recognized that the QM-MM boundary is not the ideal C-C type (and best avoided). We can override this, however, if we want by specifying the unusualboundary=True option (to QMMMTheory object). Note that ASH also prints the QM-region coordinates in the output (without any linkatoms) which can be useful to check in a visualization program to make sure that the QM-region is defined as intended.
If we use the unusualboundary=True keyword to override the ASH-exit, ASH will continue and it will automatically determine linkatoms to terminate the boundary to make 4 Fe-S-L(H) junctions. The linkatom (L) would be a H-atom added automatically by ASH but only when the QM-calculation is being performed. The QM-code would thus see coordinate for an Fe(SH)4 complex. If the QM-code is ORCA the ORCA inputfile created by ASH will look something like this:
! r2SCAN-3c tightscf
%pointcharges "orca.pc"
*xyz -1 6
S 28.35 36.955 29.371
S 32.028 37.583 29.84
S 30.309 34.16 30.681
S 30.112 37.065 32.956
Fe 30.202 36.484 30.727
H 28.14714878591415 36.3121362117398 28.51445021470574
H 32.19930481797761 38.5935834563914 29.469222793303494
H 30.094505037372087 33.61119681224084 31.598009799354752
H 30.663321996844005 37.47906579137972 33.80021180766738
*
We can see that ASH has added 4 H-atoms to the QM-region (to terminate the boundary) which allows a realistic QM-calculation to be carried out. Visualizing the coordinates we see the 4 H-linkatoms have been added, the S-H bonds all point in the direction of the actual S-CH2 junctions.
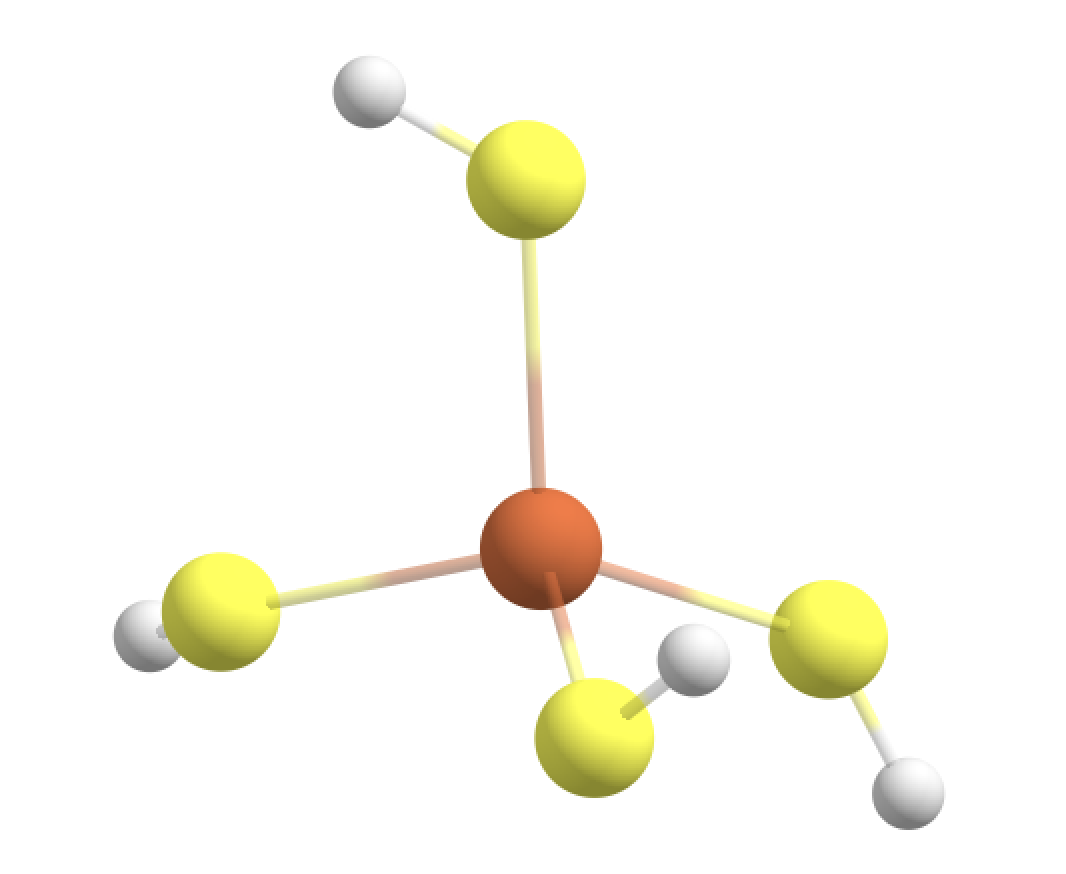
However, this type of QM-region (and resulting QM-MM boundaries) is actually bad for a few reasons:
The S-C bond is slightly polar and not the ideal C-C scenario.
Being so close to the metal ion you would risk creating artifacts. For an open-shell Fe ion you would e.g. expect some spin-density on the S-atoms and having an artificial linkatom there (during the QM-calculation) is not ideal.
For the case of the CHARMM forcefield: The S-atom is part of a charge group with the neighbouring CH2 group for both neutral CYS residues and also deprotonated CYS residues (either labelled CSD or CYM in forcefield files). A chargegroup is a group of close atoms together whose pointcharges sum to either zero (e.g. for neutral sidechains) or -1/+1 (e.g. for charged sidechains). Making a QM-MM boundary here (which sets the pointcharges of the QM-atoms to zero will create a charged fractional MM-pointcharge-group here.
Option 2: Fe, SCH2 groups in the QM-region (GOOD)
A more realistic QM-region would include the methylene (CH2) groups of each Cys residue as well.
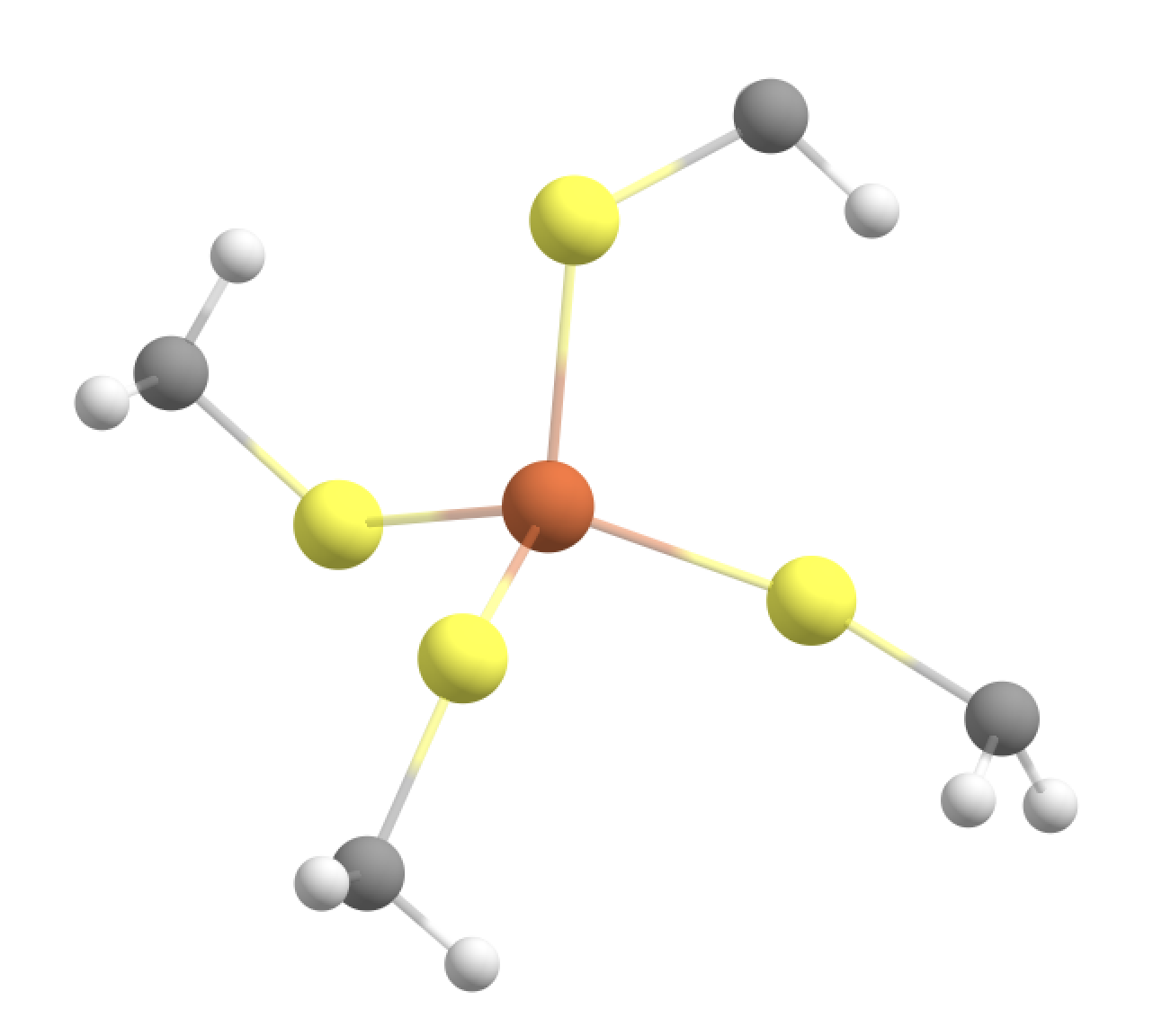
qmatoms= [93,94,95,96,133,134,135,136,564,565,566,567,604,605,606,607,755]
#Note: unusualboundary=True keyword is no longer necessary in QMMMTheory object
qmmm = QMMMTheory(qm_theory=orca, mm_theory=omm, fragment=fragment, qm_charge=-1, qm_mult=6, qmatoms=qmatoms)
This would terminate the boundary to make a Fe-S-CH2-L junction. The linkatom (L) would be an H-atom added automatically by ASH but only when the QM-calculation is being performed.
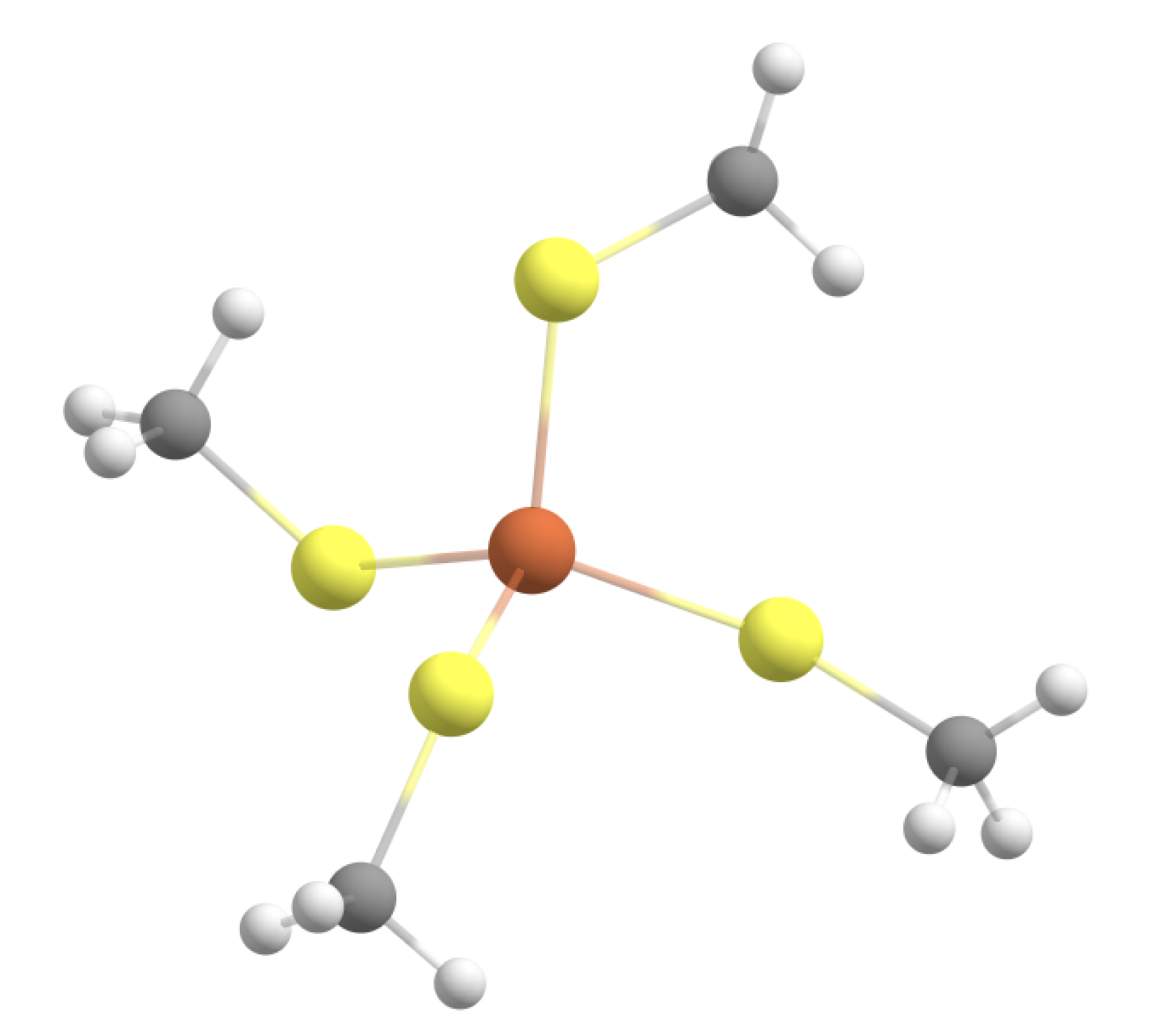
The ORCA inputfile created by ASH would look like this:
! r2SCAN-3c tightscf
%pointcharges "orca.pc"
*xyz -1 6
C 28.013 35.887 27.948
H 28.865 36.091 27.265
H 28.239 34.876 28.349
S 28.35 36.955 29.371
C 32.326 39.341 29.195
H 32.491 39.979 30.089
H 33.302 39.355 28.664
S 32.028 37.583 29.84
C 29.942 33.221 32.25
H 28.838 33.268 32.37
H 30.163 33.89 33.109
S 30.309 34.16 30.681
C 31.072 37.786 34.426
H 30.458 38.511 35.002
H 31.94 38.377 34.063
S 30.112 37.065 32.956
Fe 30.202 36.484 30.727
H 27.096022970908088 35.92119472939536 27.359711084075737
H 31.554800155167054 39.75364007067431 28.544554789852715
H 30.311808749506998 32.20547050428352 32.39156741192065
H 31.58959570776056 37.14146008749057 35.13646673707513
*
This is a good option because:
We cut through a relatively nonpolar C-C bond
We are farther away from the metal ion
We don‘t cut through the (SCH2) charge group.
This QM-region definition actually includes the whole sidechain of each CYS residue. This is usually a pretty good QM-MM boundary option for most amino acids, things get more difficult as we go beyond this as next we have to cut through a peptide backbone. Making cuts through peptide backbones is a bit more problematic. The best choice is often to avoid it if you think the QM-region is large enough but if you think it is necessary then continue below.
Option 3: Fe, SCH2 groups + Cys-6 C(alpha) + H(alpha) in the QM-region (NOT ALLOWED)
Going beyond the Cys sidechain we go into the peptide backbone, the smallest imaginable QM-region addition would involve adding the CH group which is the alpha-carbon group. We can try to do this for one of the Cys sidechains (resid 6).
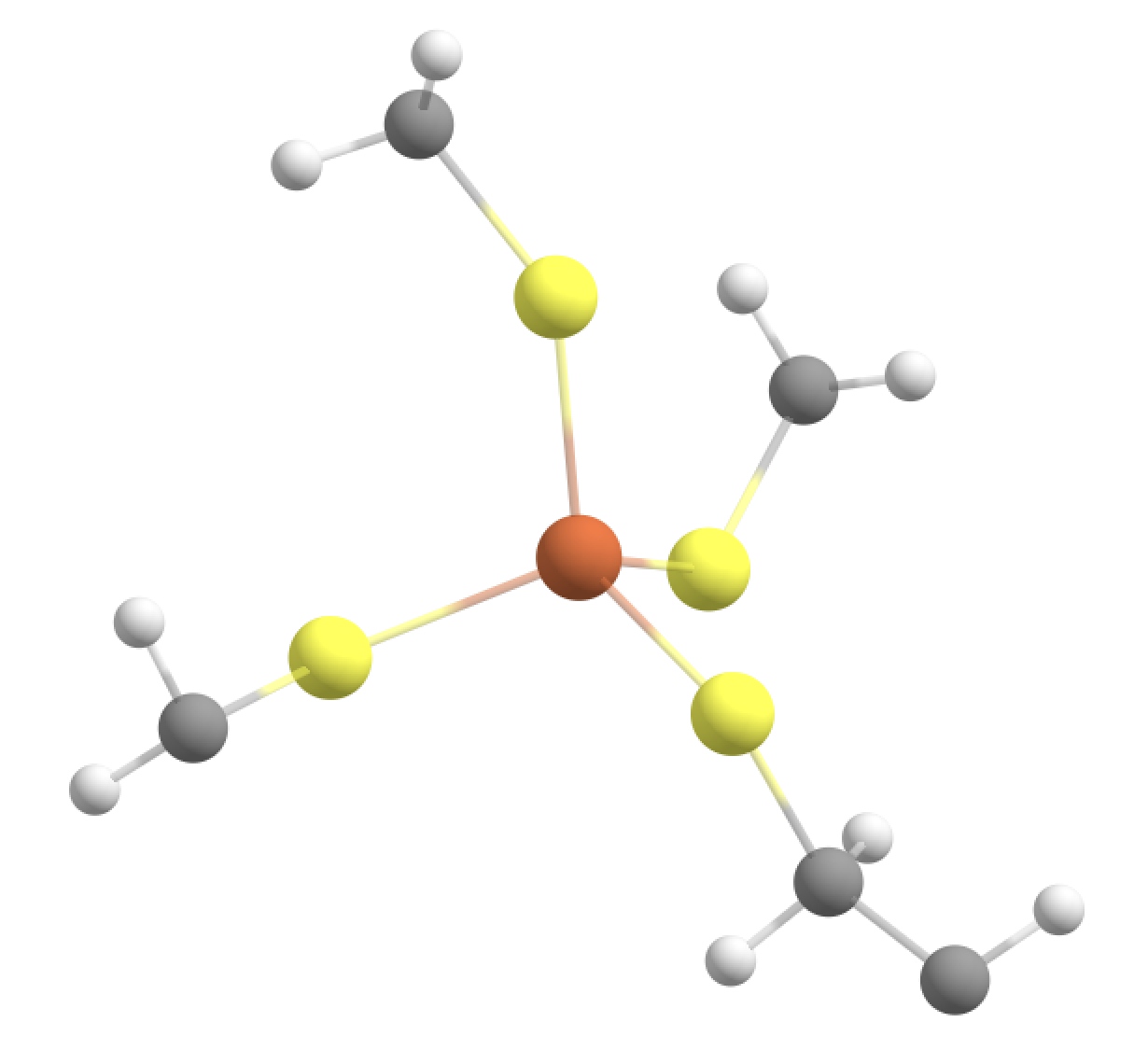
#QM-III (bad): QM-II + C_alpha and H_alpha of Cys-6 backbone. ASH will not allow this
#Adding indices 89 and 90 to the QM-region (C-alpha and H-alpha of CYS resid 6)
qmatoms= [93,94,95,96,133,134,135,136,564,565,566,567,604,605,606,607,755,89,90]
qmmm = QMMMTheory(qm_theory=orca, mm_theory=omm, fragment=fragment, qm_charge=-1, qm_mult=6, qmatoms=qmatoms)
This, however, results in ASH exiting with an error:
Determining QM-MM boundary
Parameters determing connectivity:
Scaling factor: 1.0
Tolerance: 0.4
QM atoms: [89, 90, 93, 94, 95, 96, 133, 134, 135, 136, 564, 565, 566, 567, 604, 605, 606, 607, 755]
QM atoms to be excluded from boundary creation (excludeboundaryatomlist): []
Problem. Found more than 1 boundaryatom for QM-atom 89 . This is not allowed
This typically either happens when your QM-region is badly defined or a QM-atom is clashing with an MM atom
QM atom : 89
MM Boundaryatoms (connected to QM-atom based on distance) : [87, 91]
Please define the QM-region so that only 1 linkatom would be required.
MM Boundary atom coordinates (for debugging):
87 N 26.853 34.952 26.065
91 C 26.287 37.344 26.68
ASH exiting with code: 1
This QM-region is actually not allowed by ASH as it would require adding 2 linkatoms being added to the alpha-carbon (index 89) due to 2 cuts. Furthermore for this QM-region definition we would cut through a C-N bond anyway which is not good (we would get problematic forces at the boundary).
Option 4: Fe, SCH2 groups + Cys-6 1 CH(alpha) + Cys-6 NH group + CO of Val-5 (GOOD)
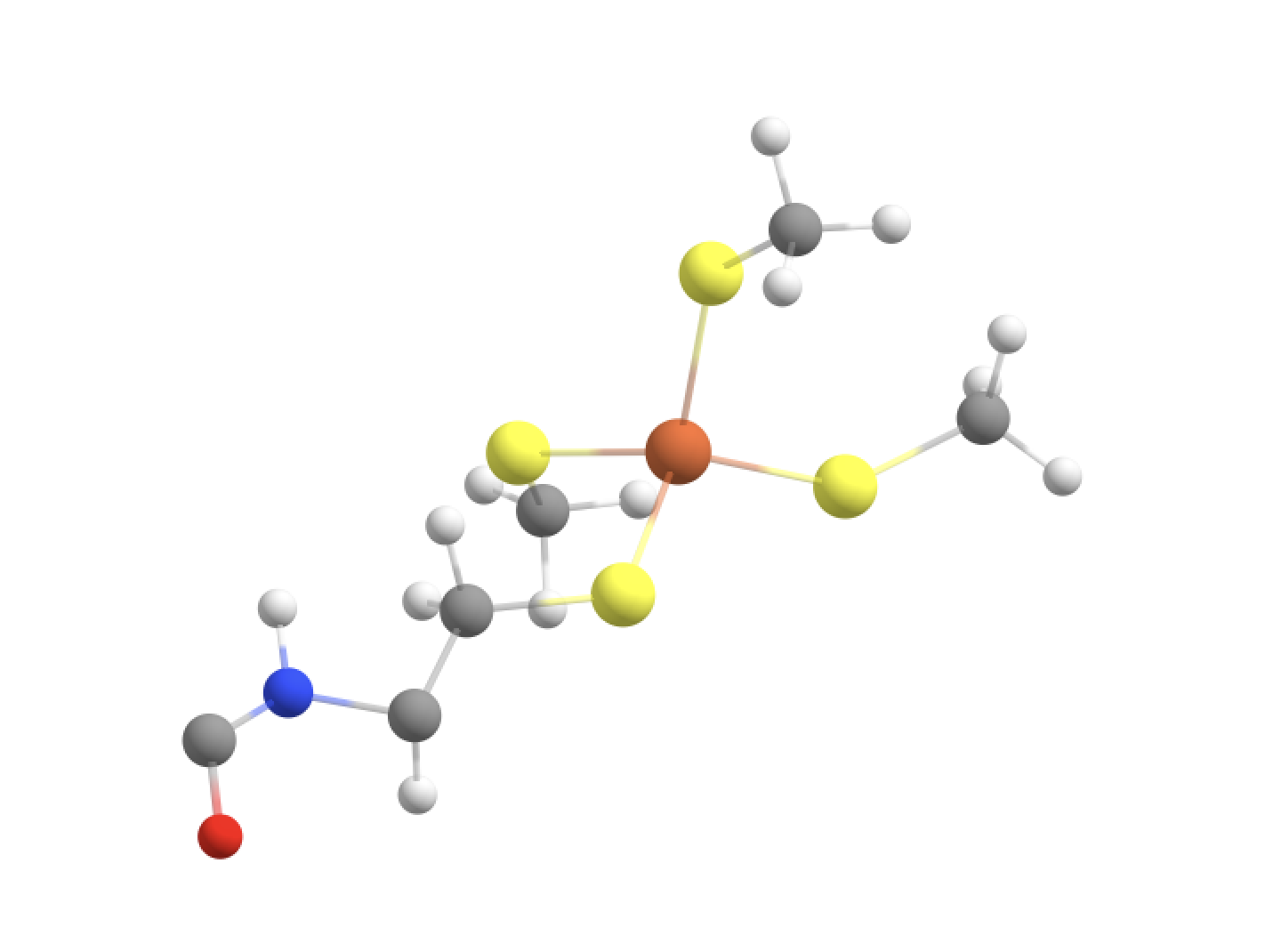
#QM-IV: QM-II + CH(alpha) and NH of Cys-6 backbone + CO of Val-5 backbone
qmatoms= [93,94,95,96,133,134,135,136,564,565,566,567,604,605,606,607,755,89,90,87,88,75,76]
qmmm = QMMMTheory(qm_theory=orca, mm_theory=omm, fragment=fragment, qm_charge=-1, qm_mult=6, qmatoms=qmatoms)
In order to avoid the double linkatom scenario (on the same QM-atom) we have to extend QM-region a little further. We are forced to include the whole nearest amide group (C=O-NH) as peptide bonds are known to have partial double-bond character and we want to avoid cutting through a C-N bond anyway.
We then end up with a dangling C=O bond on the left side of the image (belong to Val-5) where a linkatom would be added to create an effective aldehyde group here. That link replaces the C-alpha of Val-5. This cut is not perfect as the cut C-C bond involves a slightly polar carbon of the Val-5 carbonyl group but it is really the only good possibility here.
The C=O is its own CHARMM charge group (of the Val residue) so we are not cutting through a charge group (which is good). Furthermore CH(alpha) and NH together (belonging to Cys-6) also belong to a charge group but there is no problem since we are including the whole charge group.
Finally this QM-region definition also makes a cut between the Cys-6 alpha-carbon and the C of the Cys-6 C=O group. However, this is fine since now there is only 1 possible linkatom on the Cys-6 alpha-carbon. QM-region with linkatoms is shown below.
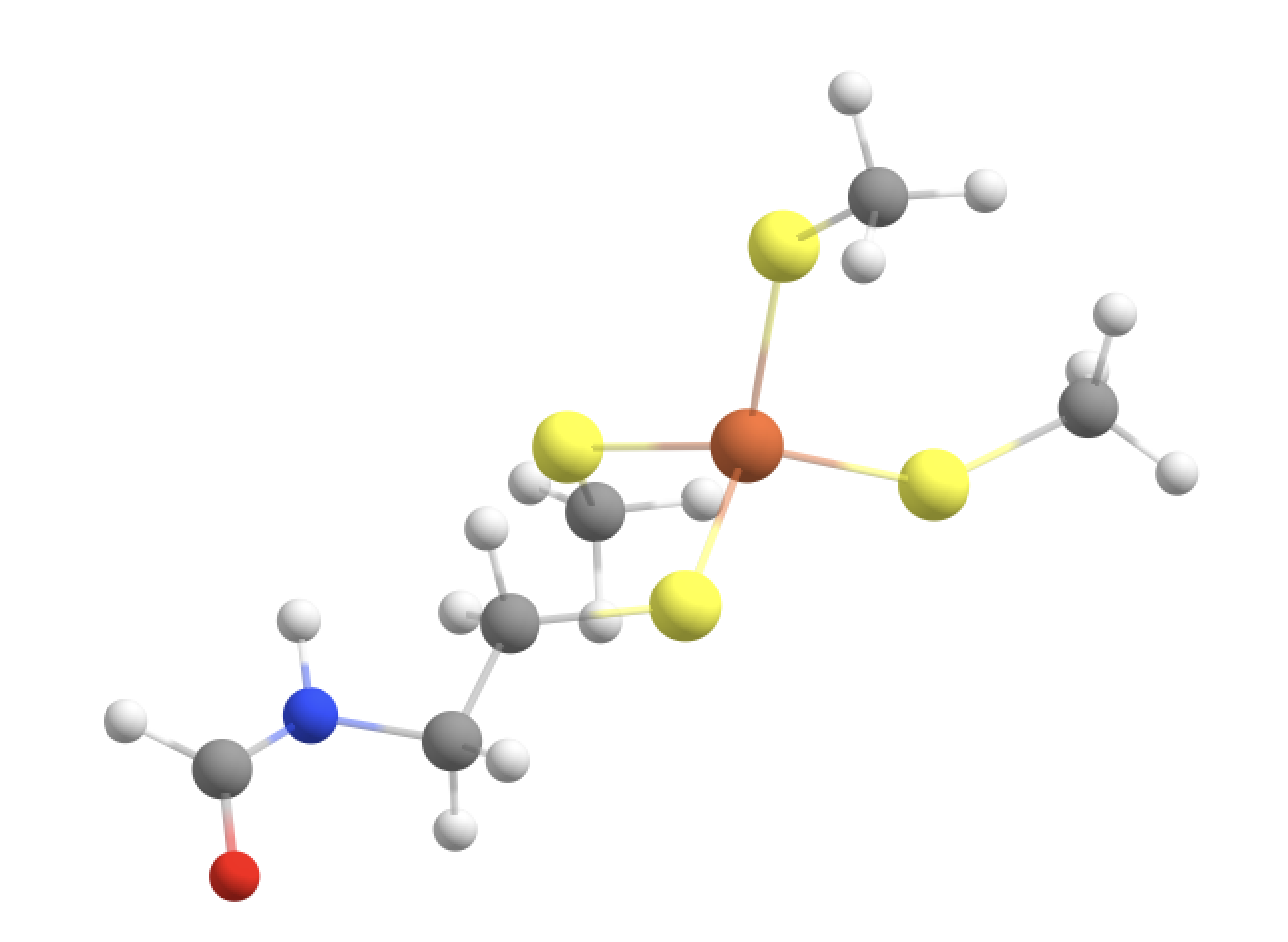
Both QM-MM boundaries here actually cut the same type of C(alpha)-C (of C=O) which is the best possible choice when cutting through a peptide backbone.
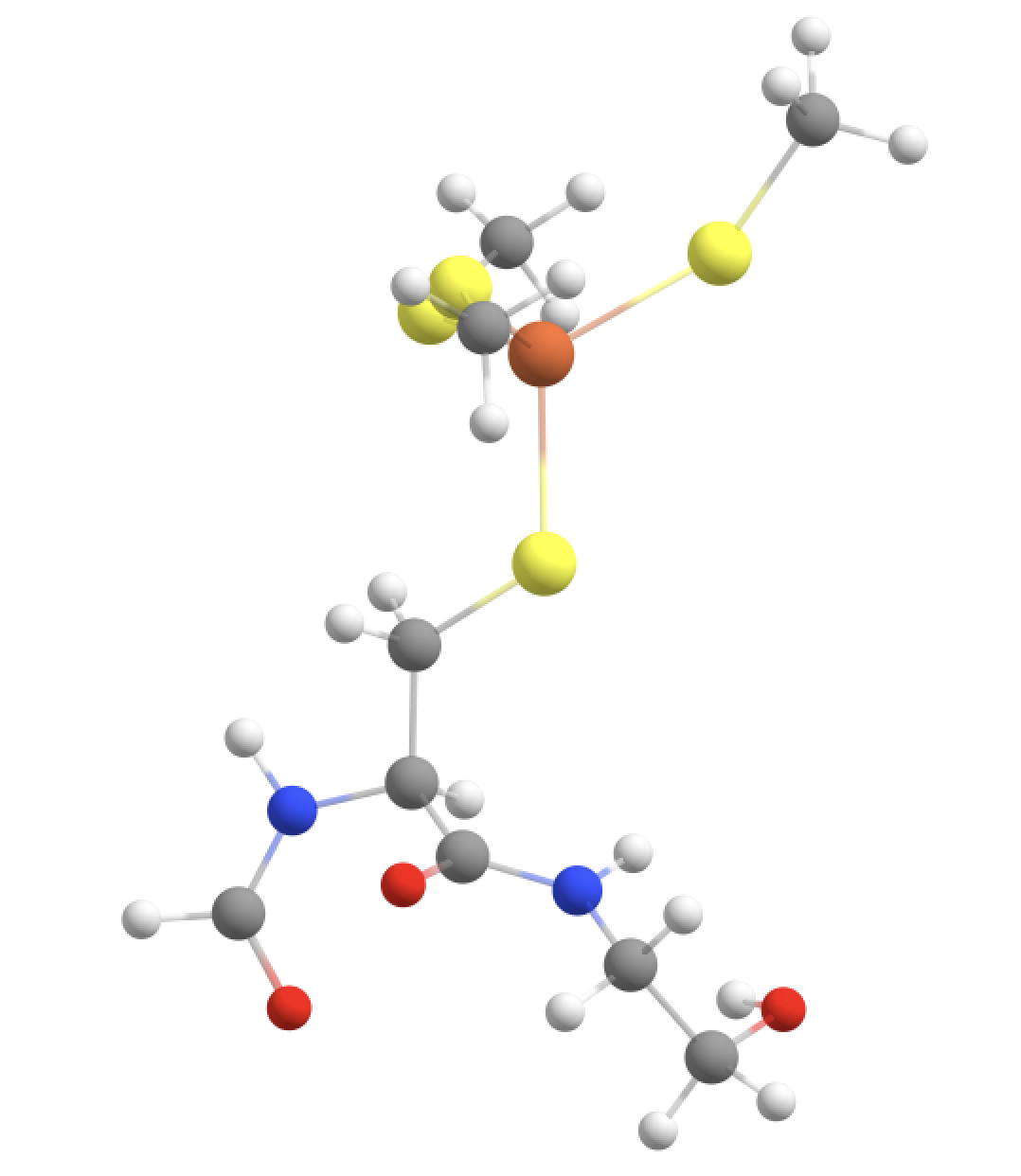
Option 5: Fe, SCH2 groups + Cys-6 CH(alpha) and NH peptide group + CO of Val-5 + NH-CH backbone of Thr-7 and part of sidechain of (GOOD)
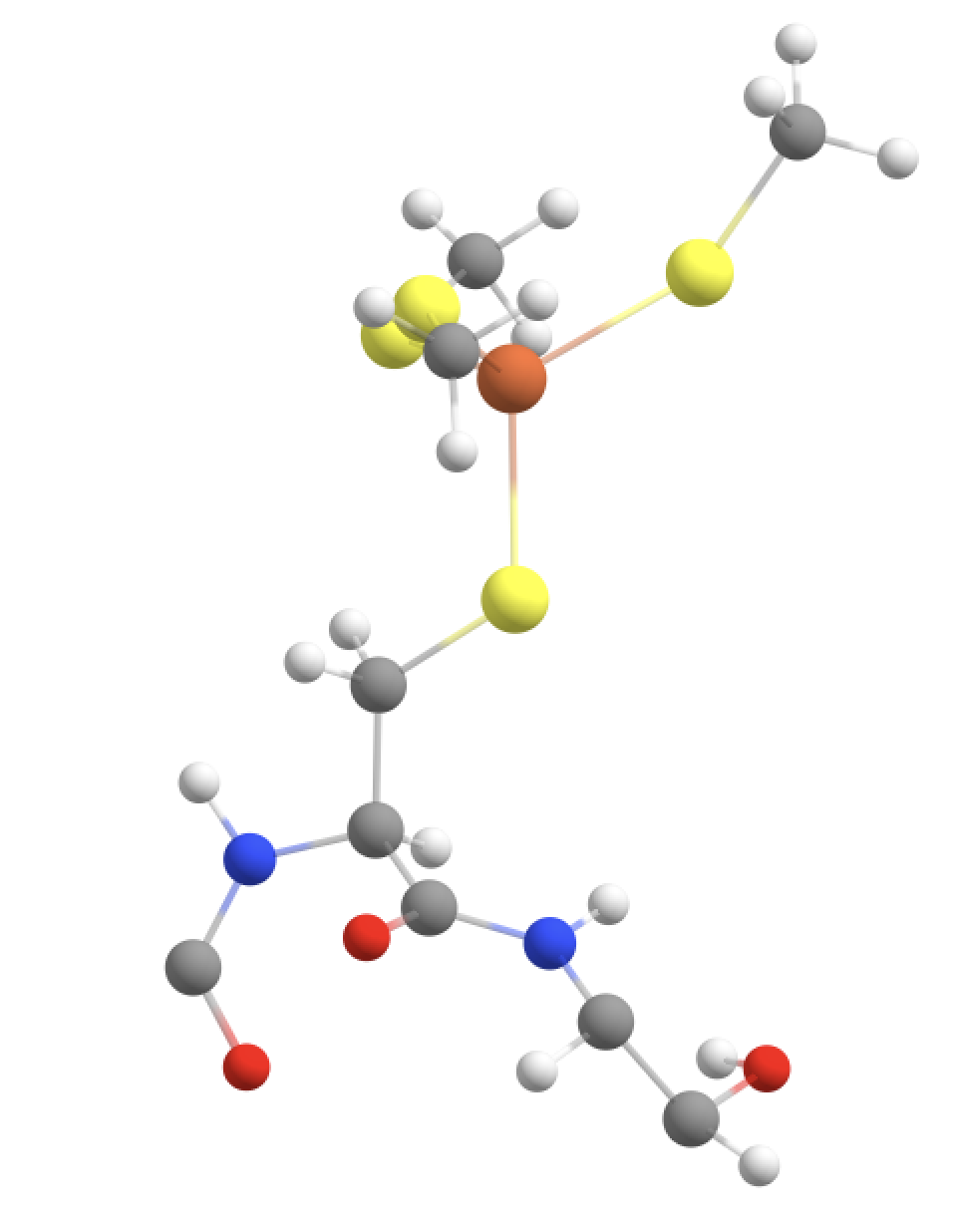
The QM-region option 4 is arguably a bit unbalanced from a QM perspective since we are only extending the peptide backbone in 1 direction. We could try to extend the QM-region further on the right side (towards Thr-7), keeping in mind that we don‘t want to cut through either the C-N bond (between Cys-6 and Thr-7) or the N-C bond (within Thr-7). Again we are forced to avoid the double-linkatom-on-same-QM-atom scenario so we have to extend further. Here we choose to include part of the sidechain of Thr-7, specifically the CH-OH part which according to the CHARMM definition of the Thr residue forms a charge group.
#QM-V: QM-IV + CH-NH of Thr-7 backbone + part of Thr-7 sidechain
qmatoms= [93,94,95,96,133,134,135,136,564,565,566,567,604,605,606,607,755,89,90,87,88,75,76,91,92,97,98,99,100,103,104,105,106]
qmmm = QMMMTheory(qm_theory=orca, mm_theory=omm, fragment=fragment, qm_charge=-1, qm_mult=6, qmatoms=qmatoms)
This now results in 3 QM-MM boundaries and 3 linkatoms are required.
Additional options:
These are not the only options for extended QM-regions. A cleaner option than option 5 above (only slightly more expensive) would include the whole sidechain of Thr-7 (1 extra methyl group).
Another option that avoids the Thr sidechain would have instead extended the peptide backbone towards Val-8. The latter would be more expensive since one would have to go quite far to make a good cut. However, since this option would actually include the Val-8 N-H bond in the QM-region which according to the X-ray structure makes a hydrogen bond to the sulfur atom of Cys-6 this may in some sense be a better option. Furthermore, for the rubredoxin active site one would normally want to expand the QM-region associated with the 4 cysteine residues in a balanced manner anyway and at some point including large peptide chain segments may be necessary.
Further reading
Overall defining extended QM-regions and making good cuts requires a bit of know-how and careful consideration. The best option is to start simple and then carefully expand the QM-region as needed.
Some useful reading: